By Nicolas Gambardella
Many people start in the translation business without a corresponding professional training. This is absolutely fine, and it is in fact a good way of using one’s language skills acquired either during a professional activity or a travelling life. However, as amateurs, they probably all tend to make the same mistakes. Here we list a few of them.
1) Believing that a translation job is just … translating
A translation job is much more than converting a text from a source language to a target language. Glossaries and a bit of grammar polishing would almost be sufficient for that. However, a translator must convey the “content” of the source document. That involves of course translating the words. But it also, and foremost, involves producing a text that carries the same message. And to do so requires to understand what the text is about, in details and with all its subtleties. This is why all translators have their specialities, and although most translators can do an OK job with any text in their paired languages, they really excel only within a few niches.
Conveying the proper meaning is sometimes at odd with keeping to a strict translation of the words themselves. Depending on the domain covered, one wants to massage the text to make it more readable and respect the form of the source text. With the exception of legal documents – where one must absolutely stick to the original, even if the result seems quite heavy – some sentence restructuring and expression switching is needed to make the result more palatable, and also truly equivalent in the target language. Finally, in the artistic domain, one wants to respect the style of the original, terse or verbose, dull or vivid, mainstream or abstruse. Lovecraft did not write like Stephen King despite hovering in the same literature space.
2) Starting the translation immediately
In order to translate a text accurately, we cannot start the work straight away. We must read the entire text beforehand, to make sure we understand what it is about, have an idea of the specialized knowledge we might need to acquire, and what was the goal of the authors. Such a preliminary read will only marginally increase the time spent on a text. Or at least it should, otherwise we are probably not spending enough time on the job! Reading a 100 000 words book before starting the translation might seem daunting, but the required time is still far less than what we will spend accurately translating those 100 000 words. And the gain down the line in terms of translation speed and accuracy largely makes up for the extra effort. During this initial read, we should make notes of anything we do not immediately get, any word or expression we did not come across in the past, and make sure we do fully understand it.
3) Trusting machine translation
Machine translation has seen astounding progress in the past few years. Software such as the Google Neural Machine Translation and (even more) DeepL , really transformed the activity to a point that, in many cases, the result really sounds like it has been produced by a native speaker, but is also better than a translation made by a casual translator, i.e. someone who would make most of the errors listed here … (By the way, this makes even more pathetic the ridiculous translations used in some places such as Stansted airport. It beggars belief that nowadays people produced voice announcements that barely make sense, and even check-in machines that speak some nonsense languages using random words assembled in sentences with no grammar whatsoever).
However, machine translation is still mostly good for straight texts, without nuance, technical jargon,
That said, machine translation is generally a good feeder for Computer Assisted Translation, which brings us to the next mistake.
4) Blindly trusting the segment-based text proposed by our CAT software
Computer Assisted Translation speeds up translation massively. It saves all the time spent translating and typing trivial pieces of text such as “the red car”, “his name was Joe” and “the sky was gray and it was likely to rain”. However, CAT cannot be trusted blindly. CAT translation is based on segmentation. The text is split in small parts, containing one or a few sentences. The software then suggest translations for each segment.
Firstly, some of those translations might come from machine translation, e.g. Google Translate or DeepL. Thus, see point 3. But very often the translations come from Translation Memories. Translation memories come with their own problems. Sometimes the translations proposed are plainly wrong, with missing words or wrong sentence parsing (resulting in wrong adjective associations for adjectives or verbs for instance). Another important issue is error propagation. If a segment was badly translated once, and this translation was recorded in TMs, it will be proposed in future translations.
A very important issue is the fact that the translations proposed for a segment is done purely on this segment, independently of the content of other segments of the text. There is rarely enough context in a single segment to discriminate between different meanings of a term.
Finally, the segmentation largely follows the punctuation in the source language. Depending on the translation, for instance in literary works where one needs to keep a style and rhythm, the optimal split might be different in the target language. Fortunately, CAT tools offer segment split/merge facilities.
5) Assuming the source document is right
This is a thorny issue. The basic position is that the source language document is correct, and we need to faithfully translate it. But this is not necessarily the case. Everyone makes mistakes, even the most thorough writers. Some mistakes are easy to spot and to correct, and many should not affect the translation, such as unambiguous spelling errors. However, others will be much harder to detect. For instance, words with similar pronunciations in English (the ubiquitous “complimentary” for “complementary”, “add” for “had”, “your” for “you’re” or the dreadful “of” for “have”), or absence of accents (or incorrect ones) in French, will lead to completely wrong translations. In many case, the context will provide a quick answer, but sometimes a bit more brain juice is needed. We should always double check that we understood the text correctly, and that our chosen translation is the only one.
Finally, horror, some “errors” are made on purpose, for stylistic reasons. In the case of a novel or a play, wrong grammar or vocabulary might be part of the plot or a defining feature of a character. In that case, we probably must provide a translation that contain a correct equivalent of the initial erroneous text …
6) Forgetting to double check the punctuation
OK, that might actually be a specific version of the previous error. Translators are linguists, and as all linguists, we are in love with punctuation (aren’t we?). Is there anything that beats the Oxford comma as a favorite topic for conversation? (except perhaps split infinitives) Surprisingly enough, this is not the case of every person, or even every writer. Punctuation can be a
“an
What is the “Brett character”? (enlightened disciples of Bacchus, lower your hand). Is it the positive fruity and floral wine aromas? Or is it the
“an off-flavour affecting negatively the positive fruity and floral wine aromas, known as “Brett character”.”
But let’s not add punctuation to Guillaume
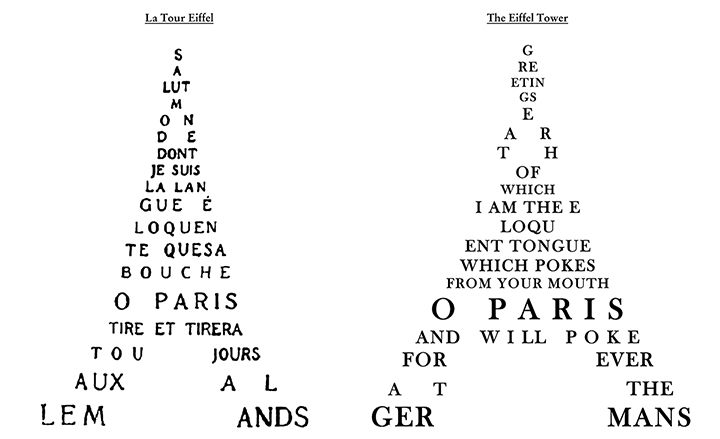
7) Not paying attention to the mainstream use bias
This error is often a side-effect of using CAT tools with TMs or MT. The proposed translations will often rely on the most frequent meaning of a term, and its most frequent translation. This is not necessarily the meaning which is the right one, or the best one, for the current source document.
Sometimes, this is just irritating. For instance, in a literary text talking about “petits détours”, CAT will keep suggesting “small detours”. While this is correct, it does not fully convey the idea carried by “petits” here. It is too bland too quantitative, and “little detours” is the best translation, as shown here, here and here.
However, the mistake can be more severe. Google Translate tells us the story of a dreadful mum, “She put a bow in her daughter’s hair” being translated into “Elle a mis un arc dans les cheveux de sa fille”. That must have hurt terribly. As was the case for the poor lad who “entered a ball” and ended up “entré dans un ballon” (GT) or even “entré dans une balle” (DeepL), instead of “entré dans un bal”. Not much room to dance there. Sometimes, the mainstream use is actually overridden by the politically correct one, and the saucy “he was nibbling at her tit” is translated into “il mordillait sa mésange”. Except if we are talking about a cat, that is a disturbing image instead of a titillating one. While those examples were a bit joky, some cases are harder to spot. Someone who planted “Indian flags” in their garden will almost always end up in French exhibiting their nationalism rather than their love of irises.
In some cases, the various meanings
8) Trying to stick 100% to the words of the source text
The true meaning of a word goes beyond its definition in a thesaurus. They carry different weight in different languages. The rude word meaning faeces is used as an interjection in almost every language. However, the level of rudeness is different in all western European countries, and sometimes choosing another rude word of the adequate level is better (no, we will not provide examples). And of course, there are very few cases where anyone should translate “it rains cats and dogs” into “il pleut des chats et des chiens”. One should always translate it into “il pleut comme vache qui pisse” (it rains as if a cow was pissing). While the new image is no so much better, at least no animal is hurt.
9) Trying to stick 100% to the structure of the source text
Trying to reproduce absolutely the structure of the source document is very tempting and encouraged by the segmentation process of CAT tools. However, this is lazy. English sentences are known to be shorter than French ones. Therefore, translating a sentence from the latter language might require several in the former. Let’s not speak of German where an entire sentence might end up in a single word! As usual, first comes the meaning, then the rhythm, then the style. Not only this requires to merge/split sentences, it might also require swapping propositions or sentences.
10) Not reading back the complete resulting translation
Last but not least, we should never forget to re-read attentively the entire translation. In the profession, proofreading is often mentioned as an activity disconnected from translation. But no translation work should be considered complete without a proofreading step! This is even more important if CAT software were used. They are known to promote “sentence salads”, where heterogeneous texts, in style and vocabulary, are caused by using the memory of many previous translations.
What about yourself? Which mistake did you make when learning how to become an accurate and efficient translator?