By Nicolas Gambardella
You are about to embark on a system biology project which will involve some modelling. Here are a few tips to make this adventure more productive and pleasant.
1 – Think ahead
Do not start building the model without knowing where you are going. What do you want to achieve by building this model? Is it only a quick exercise, a one-off? Or do you want this model to become an important part of your current and future projects? Will the model evolve with your questions and the data you acquire? A model with a handful of variables, created to explore an idea quickly, and a model that will be parameterised with experimental measurements, whose predictions will be tested and that will be further expanded are two completely different beasts. Following the 9 tips below in the former case is
2- Focus on the biology
A good systems biology model aims to be anchored in biological knowledge and even (generally) reflects the system’s behaviours’ biological mechanisms. We are using modelling to understand biology and not using biology to illustrate modelling techniques (which is a perfectly respectable activity but not the focus of this blog post). In order to do so, the model must be built from the processes we want to represent (hence complying with the Minimum Information Requested in the Annotation of Models). Therefore, try to build up your model from reactions (or transitions if this is a Petri Net, rules for a Rule-based model, influences for a Logic model), rather than writing directly the equations controlling the evolution of variables.
Another aspect worth a thought is the existence of different “compartments”. In systems biology, compartments are the “spaces” that contain the biological entities represented by your variables (the word has a slightly different meaning in PKPD modelling, meaning the variable itself). Because compartments can have different sizes, these sizes can change, and they can be used to affect other aspects of the models, it is important to represent them correctly, rather than ignoring them altogether, which was the case for decades.
Many tools have been developed to help you build models that way, such as (but absolutely not limited to) CellDesigner and the excellent COPASI. These software tools are, in general, very user-friendly and more approachable for biologists. An extensive list of tools is available from the SBML software guide.
3- Document as you build
Bookkeeping is a cornerstone of any professional activity, and lab notebooks are scientists’ best friends. Modelling is no exception. If you do not log why you created a variable or a reaction, what biological entities they represent, how you chose the initial values or the boundaries for parameter estimation, you will make your life down the line hell. You will not be able to interpret the results of simulations, modify the model, share it with collaborators, write a publication, etc. You must start this documentation as soon as you begin building the model. Memory fades quickly, and motivation even quicker. The biggest self-delusion (or plain lie) is “I am efficient and focused now, and I must get results done. I will clean up and document the model later.” You will most probably never clean up and document the model. And if you do, you will suffer greatly, trying to remember why the heck you made those choices before.
Several software tools, such as COPASI, provide means of annotating every single element of a model, either with free
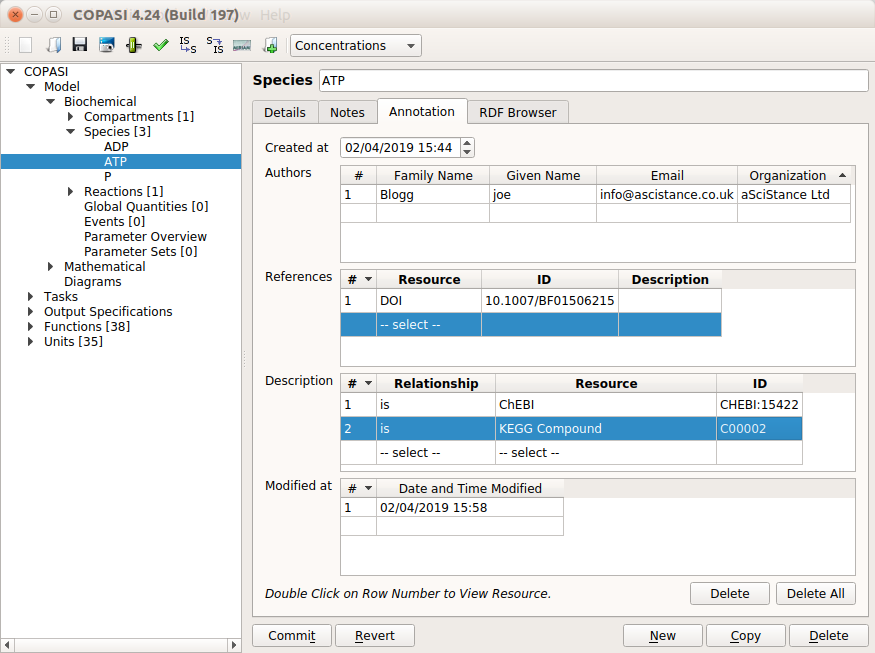
4- Choose a consistent naming scheme
This sounds like a mundane concern. But it is not! Variable and parameter names are the first layer of documentation (see tip 3). It also anchors your model in biology (tip 2). A naming scheme that is logical and consistent while easy to remember and use will also greatly facilitate future extensions of your model (tip 1). NB: we do not want to open a debate “identifiers versus accession number versus usable name” or the pros and cons of semantics in identifiers (see the paper by McMurry et al for a great discussion on that topic). Here, we talk about the short names one sees in equations, model graphs, etc.
Avoid very long names if not needed (“adenosine triphosphate”), but do not be over-parsimonious (“a”). “
5- Choose granularity and independent variables wisely
We often make two mistakes when describing systems in biology mathematically. The first one is a variant of the “spherical cow“. In order to facilitate the manipulation of the model, it is very tempting to create
The second, mirroring,
It is therefore paramount to choose the right level of granularity. There is no universal and straightforward solution, and we can encounter extreme cases. d’Alcantara et al 2003 represented calmodulin is represented by two variables (total concentration and concentration of active molecules). In Stefan et al 2008, calmodulin is represented by 96 variables (all calcium-binding combinations plus binding to other proteins and different structural conformations). Nevertheless, both papers study the biological phenomenon.
The right answer is to pick the variable granularity depending on the questions asked and the data available. A rule of thumb is to start with a small number of variables that can be matched (directly or via mathematical transformations) with the quantities you have measurements for. Then you can progressively make your model more complex and expressive as
6- Create your relationships
Once you have defined your variables, you can create the necessary relationships, which are all the mathematical constructs that link variables and parameters together. Graphical software such as CellDesigner or GINsim permit to draw the diagrams representing the processes or the influences respectively.
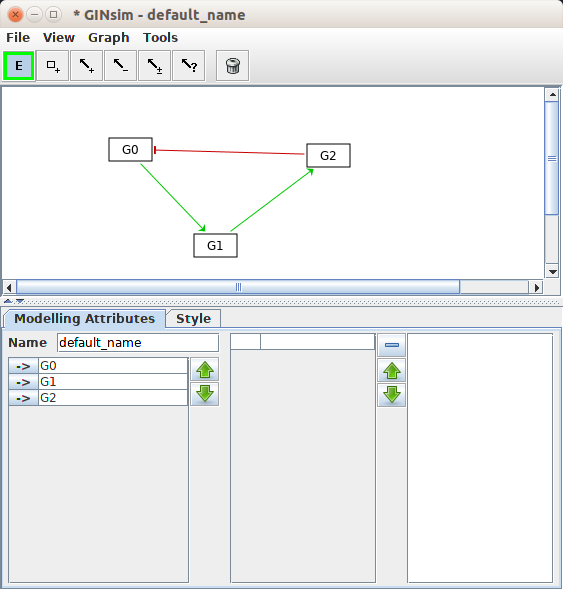
Note that some software tools provide shorthand notations that permit the creation of variables and parameters directly when writing the reactions. This is very handy for creating small models instantly. However, I would refrain from doing so if you want to document your model properly (it also makes it easier to create spurious variables and “dangling ends” through typos in the variable names).
Working on the relationships after defining the variables also easily permits the model’s modification. For example, you can add or remove a reaction without having to go through the entire model as you would with a list of ordinary differential equations.
7- Choose your math
The beauty of mathematical models is that you can explore a large diversity of possible linkages between molecular species, actual mechanisms hidden behind the “arrow” representing a process. A transformation of X in a compartment into Y in another compartment can be controlled for instance by a constant flux (don’t do that!), a passive diffusion, a rate-limited transport, or even exotic higher-order kinetics. At that point, we could write: [insert clone of tip 5 here]. Indeed, while the mathematical expressions you choose can be arbitrarily complex, the more parameters you have, the harder it will be to find proper values for them.
If the model is carefully designed, switching between kinetics should not be too difficult. A useful habit to take is to preferentially use global parameters (which scope is the entire model/module) rather than parameters defined for a given reaction/mathematical expression. Doing so will, of course, ease the use of the parameter in different expressions and facilitate the documentation and ease future model extensions, for instance, where a parameter no longer has a fixed value but is affected by other things happening in the model.
8- Plug holes and check for mistakes
Now that you have your shiny model, you need to make sure you did not forget to close a porthole that would sink it. Do you have rate laws generating negative concentrations? Conversely, does your model generate umpteen amounts of certain molecules which are not consumed, resulting in preposterous concentrations? Software like COPASI have checks for this kind of thing. In the example below, I created a reaction that consumes ATP to produce ADP and P, with a constant flux. This would result in infinite concentrations of ADP and infinitely negative concentrations of ATP. COPASI catches it, albeit returning a message that could be clearer.
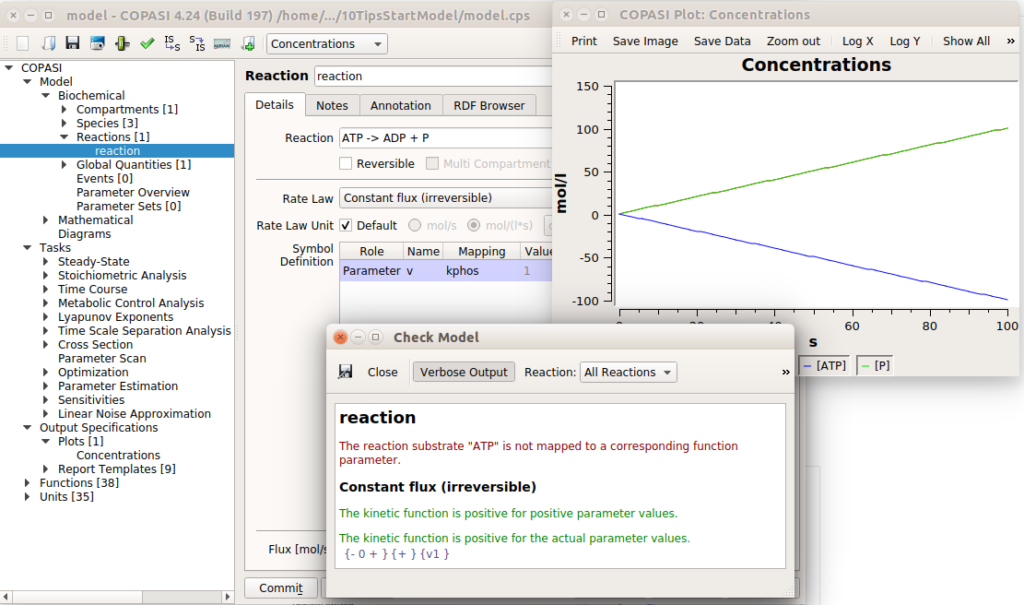
Ideally, a model should be “homeostatic”. All molecular species should be produced and consumed. Pure “inputs” should be produced by creation/import reactions, while pure “outputs” should be consumed by degradation/export reactions. Simulating the model would not lead to any timecourse tending to either +∞ or -∞.
9- Create output
“A picture is worth a thousand words”, and the impact of the results you obtained with such a nice will be greater if served in clear, attractive and expressive figures. Timecourses are useful. But they are not always the best way to present the key message. You want to show the effect of parameter values on molecular species’ steady-states? Try parameter scanning plots, and their derivatives, such as bifurcation plots. Try phase-portraits. Distributions of concentrations during stochastic simulations or after ensemble simulations can be represented with histograms. And why being limited to 2D-plots? Use 3D plots and surfaces instead, possibly in conjunction with interactive display (plot.ly …).
10- Save your work!
Finally, and this is quite important, save often and save all versions. Models are code, and code must be versioned. You never know when you will realise you made a mistake and will want to go back a few steps and start exploring a different direction. You certainly do not want to start all over again. Recent work explored ways of comparing model versions (see the works from the Waltemath group, for instance). But we are still some way off the possibility of accurately “diff and merge” as it is done on text and programming code. The safest way is to save all the significant versions of a model separately.